tagtango
tagtango.RmdInput data requirements
tagtango is flexible in handling various data types. To
date, it accepts input in the form of either a
MultiAssayExperiment R object stored as an RDS file (Ramos et. al.,
2017), a SingleCellExperiment R object stored as an RDS
file (Amezquita et.
al., 2020), or an R data frame stored as an RDS, CSV, or TSV
file.
MultiAssayExperiment object
Providing a MultiAssayExperiment object as input will
allow you to study multiple data modalities simultaneously. However,
there are certain criteria that needs to be met. First, the elements of
the ExperimentList container should be
SingleCellExperiment following the specifications stated in
the next section. Second, cells in all elements of the
ExperimentList should be the same (and have unique and
matching names). Finally, different annotations should be stored as
columns of the colData data frame within the object.
As test dataset, a preprocessed and annotated 10x dataset is provided
with the package. This is a MultiAssayExperiment with
Peripheral Blood Mononuclear Cells (PBMCs) from a healthy donor stained
with a few TotalSeq-B antibodies (10x
Genomics, 2018), and is readily accessible via:
## A MultiAssayExperiment object of 2 listed
## experiments with user-defined names and respective classes.
## Containing an## ExperimentList class object of length 2:
## [1] RNA: SingleCellExperiment with 33538 rows and 7472 columns
## [2] ADT: SingleCellExperiment with 17 rows and 7472 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesNotice that the column and row names of each
SingleCellExperiment in the corresponding
ExperimentList object are not NULL, and that
the different annotations are stored in:
MultiAssayExperiment::colData(test_data)## DataFrame with 7472 rows and 21 columns
## Sample Barcode sum
## <character> <character> <numeric>
## AAACCCAAGATTGTGA-1 /var/folders/w9/2_9f.. AAACCCAAGATTGTGA-1 6160
## AAACCCACATCGGTTA-1 /var/folders/w9/2_9f.. AAACCCACATCGGTTA-1 6713
## AAACCCAGTACCGCGT-1 /var/folders/w9/2_9f.. AAACCCAGTACCGCGT-1 3637
## AAACCCAGTATCGAAA-1 /var/folders/w9/2_9f.. AAACCCAGTATCGAAA-1 1244
## AAACCCAGTCGTCATA-1 /var/folders/w9/2_9f.. AAACCCAGTCGTCATA-1 2611
## ... ... ... ...
## TTTGTTGGTTCAAGTC-1 /var/folders/w9/2_9f.. TTTGTTGGTTCAAGTC-1 5830
## TTTGTTGGTTGCATGT-1 /var/folders/w9/2_9f.. TTTGTTGGTTGCATGT-1 4096
## TTTGTTGGTTGCGGCT-1 /var/folders/w9/2_9f.. TTTGTTGGTTGCGGCT-1 5524
## TTTGTTGTCGAGTGAG-1 /var/folders/w9/2_9f.. TTTGTTGTCGAGTGAG-1 4039
## TTTGTTGTCGTTCAGA-1 /var/folders/w9/2_9f.. TTTGTTGTCGTTCAGA-1 4213
## detected subsets_Mito_sum subsets_Mito_detected
## <integer> <numeric> <integer>
## AAACCCAAGATTGTGA-1 2194 523 11
## AAACCCACATCGGTTA-1 2093 415 11
## AAACCCAGTACCGCGT-1 1518 287 11
## AAACCCAGTATCGAAA-1 737 110 12
## AAACCCAGTCGTCATA-1 1240 156 11
## ... ... ... ...
## TTTGTTGGTTCAAGTC-1 2178 323 11
## TTTGTTGGTTGCATGT-1 1256 256 11
## TTTGTTGGTTGCGGCT-1 1907 574 11
## TTTGTTGTCGAGTGAG-1 1605 271 10
## TTTGTTGTCGTTCAGA-1 1549 209 11
## subsets_Mito_percent altexps_Antibody.Capture_sum
## <numeric> <numeric>
## AAACCCAAGATTGTGA-1 8.49026 981
## AAACCCACATCGGTTA-1 6.18203 1475
## AAACCCAGTACCGCGT-1 7.89112 7149
## AAACCCAGTATCGAAA-1 8.84244 6831
## AAACCCAGTCGTCATA-1 5.97472 6839
## ... ... ...
## TTTGTTGGTTCAAGTC-1 5.54031 9520
## TTTGTTGGTTGCATGT-1 6.25000 7763
## TTTGTTGGTTGCGGCT-1 10.39102 1973
## TTTGTTGTCGAGTGAG-1 6.70958 4316
## TTTGTTGTCGTTCAGA-1 4.96084 5682
## altexps_Antibody.Capture_detected
## <integer>
## AAACCCAAGATTGTGA-1 17
## AAACCCACATCGGTTA-1 17
## AAACCCAGTACCGCGT-1 17
## AAACCCAGTATCGAAA-1 17
## AAACCCAGTCGTCATA-1 17
## ... ...
## TTTGTTGGTTCAAGTC-1 17
## TTTGTTGGTTGCATGT-1 17
## TTTGTTGGTTGCGGCT-1 17
## TTTGTTGTCGAGTGAG-1 17
## TTTGTTGTCGTTCAGA-1 16
## altexps_Antibody.Capture_percent total sizeFactor
## <numeric> <numeric> <numeric>
## AAACCCAAGATTGTGA-1 13.7376 7141 1.301038
## AAACCCACATCGGTTA-1 18.0142 8188 1.417836
## AAACCCAGTACCGCGT-1 66.2804 10786 0.768162
## AAACCCAGTATCGAAA-1 84.5944 8075 0.262742
## AAACCCAGTCGTCATA-1 72.3704 9450 0.551463
## ... ... ... ...
## TTTGTTGGTTCAAGTC-1 62.0195 15350 1.231340
## TTTGTTGGTTGCATGT-1 65.4608 11859 0.865106
## TTTGTTGGTTGCGGCT-1 26.3172 7497 1.166710
## TTTGTTGTCGAGTGAG-1 51.6577 8355 0.853067
## TTTGTTGTCGTTCAGA-1 57.4229 9895 0.889817
## Main.ADT Fine.ADT RNA.Azimuth.L1 RNA.Azimuth.L2
## <character> <character> <character> <character>
## AAACCCAAGATTGTGA-1 cluster 2 cluster 9 Mono CD14 Mono
## AAACCCACATCGGTTA-1 cluster 2 cluster 3 Mono CD14 Mono
## AAACCCAGTACCGCGT-1 cluster 2 cluster 3 Mono CD14 Mono
## AAACCCAGTATCGAAA-1 cluster 5 cluster 4 NK NK
## AAACCCAGTCGTCATA-1 cluster 5 cluster 4 NK NK
## ... ... ... ... ...
## TTTGTTGGTTCAAGTC-1 cluster 5 cluster 4 B B intermediate
## TTTGTTGGTTGCATGT-1 cluster 8 cluster 8 CD8 T CD8 Naive
## TTTGTTGGTTGCGGCT-1 cluster 2 cluster 3 Mono CD14 Mono
## TTTGTTGTCGAGTGAG-1 cluster 4 cluster 6 other T MAIT
## TTTGTTGTCGTTCAGA-1 cluster 4 cluster 6 CD4 T CD4 TCM
## RNA.Azimuth.L3 RNA.singleR.L1 RNA.singleR.L2
## <character> <character> <character>
## AAACCCAAGATTGTGA-1 CD14 Mono Monocytes Monocytes
## AAACCCACATCGGTTA-1 CD14 Mono Monocytes Monocytes
## AAACCCAGTACCGCGT-1 CD14 Mono Monocytes Monocytes
## AAACCCAGTATCGAAA-1 NK_1 NK cells NK cells
## AAACCCAGTCGTCATA-1 NK_1 NK cells NK cells
## ... ... ... ...
## TTTGTTGGTTCAAGTC-1 B intermediate lambda NK cells NK cells
## TTTGTTGGTTGCATGT-1 CD8 Naive CD8+ T-cells CD8+ T-cells
## TTTGTTGGTTGCGGCT-1 CD14 Mono Monocytes Monocytes
## TTTGTTGTCGAGTGAG-1 MAIT CD8+ T-cells CD4+ Tem
## TTTGTTGTCGTTCAGA-1 CD4 TCM_1 CD4+ T-cells CD4+ Tcm
## ADT.citesort ADT.citesort.rename
## <character> <character>
## AAACCCAAGATTGTGA-1 147_leaf CD45RO- CD16- monocyte
## AAACCCACATCGGTTA-1 151_leaf CD45RA+ CD56+ CD16- ..
## AAACCCAGTACCGCGT-1 125_leaf CD45RA+ CD45RO- CD16..
## AAACCCAGTATCGAAA-1 63_leaf CD16+ CD56+ NK cell
## AAACCCAGTCGTCATA-1 63_leaf CD16+ CD56+ NK cell
## ... ... ...
## TTTGTTGGTTCAAGTC-1 38_leaf CD16+ CD56+ TIGIT+ B..
## TTTGTTGGTTGCATGT-1 23_leaf naive CD8+ T cell
## TTTGTTGGTTGCGGCT-1 67_leaf CD45RA+ CD45RO- CD16..
## TTTGTTGTCGAGTGAG-1 169_leaf central memory CD4+ ..
## TTTGTTGTCGTTCAGA-1 159_leaf central memory CD4+ ..
SingleCellExperiment object
Providing a SingleCellExperiment object, one also needs
to ensure that the data is formatted in a specific manner. First, data
should be normalized and stored as a logcounts assay within
the SingleCellExperiment object. For example, in the main
text, CITE-seq data was normalized using the R package ADTnorm.
Second, cell and marker names within the
SingleCellExperiment object should be defined (ensure these
are not set as NULL). Finally, different annotations should
be stored as columns of the colData within the object.
Likewise, the object can contain the principal components of the data
calculated using different decomposition techniques in
reducedDims (see Amezquita et.
al. 2020 for further information). Notice that an example of a
SingleCellExperiment that can be used as input by
tagtango can be generated using:
test_data[["ADT"]]## class: SingleCellExperiment
## dim: 17 7472
## metadata(1): Samples
## assays(1): logcounts
## rownames(17): CD3 CD4 ... IgG1 IgG2b
## rowData names(3): ID Symbol Type
## colnames(7472): AAACCCAAGATTGTGA-1 AAACCCACATCGGTTA-1 ...
## TTTGTTGTCGAGTGAG-1 TTTGTTGTCGTTCAGA-1
## colData names(20): Sample Barcode ... RNA.singleR.L2 ADT.citesort
## reducedDimNames(1): UMAP
## mainExpName: ADT
## altExpNames(0):Data frame object
Providing a data frame object is the simplest way to run
tagtango. The application expects different annotations to
be stored as columns of the data frame. An example of a data frame that
can be used as input by tagtango can be generated again
with:
MultiAssayExperiment::colData(test_data)Supplementary usage scenario: comparing spatial transcriptomics annotations
To highlight the versatility of tagtango, we tested our
software on a different data modality. We used an annotated spatial
transcriptomics dataset provided as part of the spatialLIBD
project (Maynard
et. al., 2021;, Pardo et. al.,
2022). The data was generated with 10x Genomics Visium platform and
contain human brain tissue samples from three healthy donors. In
particular, these are spatially adjacent replicates of human
dorsolateral prefrontal cortex tissue. This dataset is interesting
because it investigates the laminar structure of the brain cortex,
providing several manual annotations of the different layers for the
individual spots (considered as `ground truth’) as well as several
cluster-based annotations.
Following the input data requirements outlined above, we prepared the
dataset with R to be studied with tagtango. First, we
followed Pardo et.
al., (2022), and downloaded the spatial transcriptomics dataset.
# Load libraries
library(spatialLIBD)
library(SingleCellExperiment)
library(scater)
# Download data
spe <- fetch_data(type = "spe")The data comes in the form of a SpatialExperiment R
object. We reshaped the data into a SingleCellExperiment
object using
sce <- SingleCellExperiment(
list( logcounts = logcounts(spe) ),
colData = cbind( colData(spe), spatialCoords(spe) ),
rowData = rowData(spe)
)
sce <- runUMAP(sce, exprs_values = "logcounts")Notice that the spatial coordinates of the spatial transcriptomics
dataset were added as columns in the colData object (these
could also be added as part of the reducedDims object), and
that we calculated the UMAP decomposition using the R package
scater (McCarthy et. al.,
2017).
Finally, this object contains multiple samples for a spatial transcriptomics dataset; therefore, the column names are not unique. Likewise, row names (i.e. gene IDs) can also be more intuitive if we use the gene names as opposed to ensembl names. To fix this, we used:
# Row names
rownames(sce) <- rowData(sce)$gene_name
# Fix col names (they were not unique)
colnames(sce) <- rownames(data.frame(colData(sce)))Using tagtango, we then compared sets of annotations for
this dataset. A first interesting comparison is that between the
consensus manual annotations and one of the best performing unsupervised
clustering method used in (Maynard et. al.,
2021). In particular, we focused on the differences between ‘ground
truth’ annotations and annotations using highly variable genes from
scran (Lun
et. al., 2016), 50 PCs for dimension reduction, and spatial
coordinates as features (i.e. method HVG\_PCA\_spatial as
described in Maynard et. al.,
2021). Figure 1 explores the comparison between spots manually
labelled as White Matter (WM) but annotated as two different clusters in
the unsupervised approach. Notice that we used tagtango to
filter out the results, focusing on one sample (i.e. labelled as
151675) excluding non-tissue spots (i.e. classified as
NA in the manual annotations). This comparison identified
key genes driving the distinction between sub-populations within the
manually annotated WM region. These include known WM and L5 marker genes
such as MBP, which, coupled with the fact that spots in cluster 4 did
not intersect with spots manually labelled as L5 or L6, suggests
additional modularity within the WM layer. Similarly, a marker gene for
gray matter/neurons that was used to define the sample orientation by Maynard et. al.,
2021, SNAP25 is also identified as relevant. Finally, MOBP, a gene
identified as top 10th most variable markers across layers by the author
but that our comparison suggests this also show some level of modularity
within WM, was also selected as relevant.
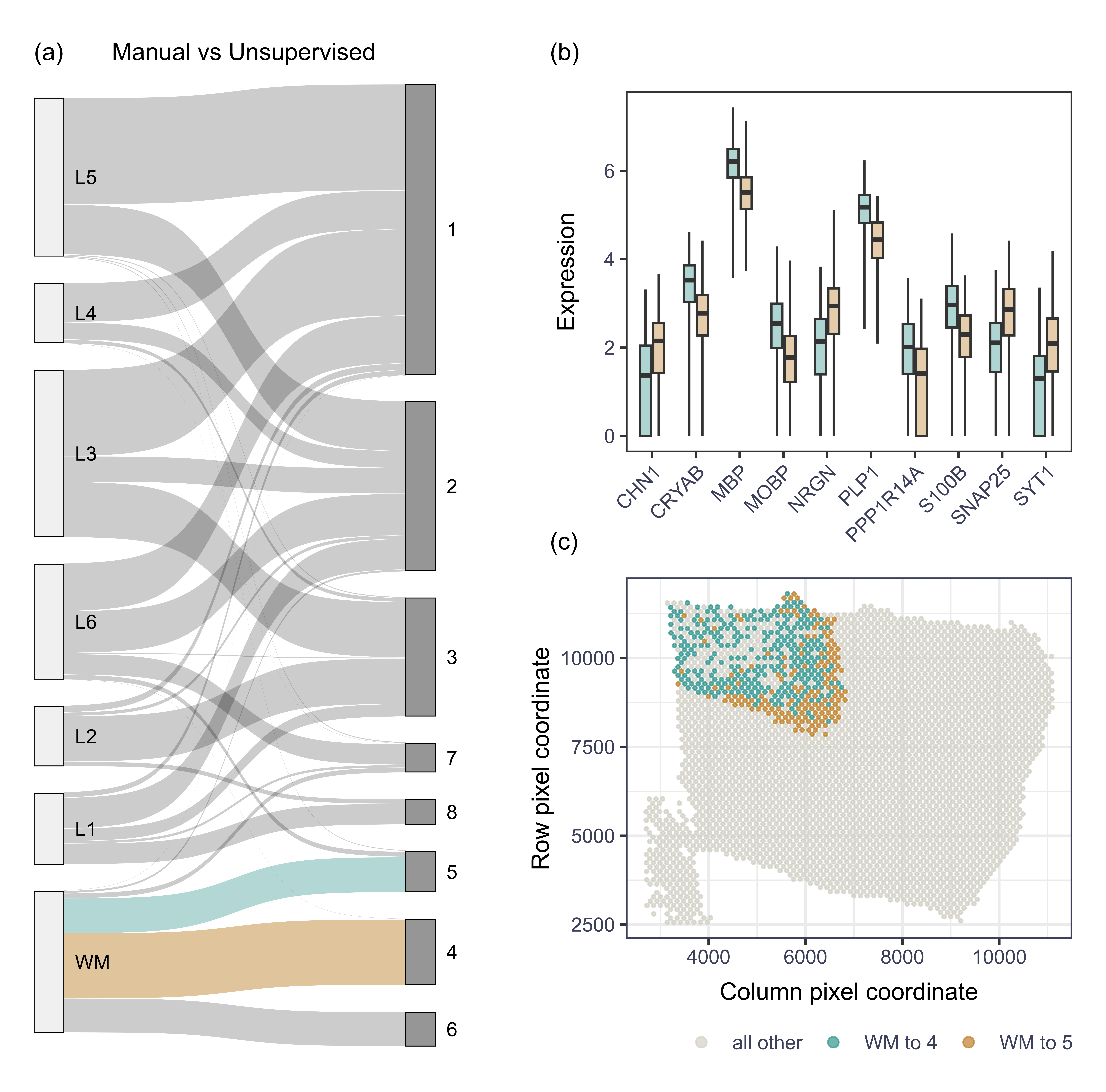
tagtango to only include cells
from sample `151675’ and in-tissue spots. The coloured links in the
diagram indicate the cell populations selected for White Matter (WM).
Panel (b) displays a direct comparison of the normalized RNA expression
for the two selected cell populations, including only markers selected
as relevant. The colours of the bars match those of the selected links
in panel (a). Panel (c) presents the spatial representation of all
spots, where the colours of the points match those of the selected links
in panel (a).Notice that there are multiple types of comparisons that could be
performed with tagtango. For example, we could try to
understand differences between the way the different co-authors of the
study labelled the spots, annotations that are also provided with the R
package. Likewise, we could focus on comparing full layers, reproducing
the authors results and potentially identifying additional marker genes
separating the cortex layers.
Supplementary usage scenario: understanding batch effects
An interesting use case for tagtango is the
identification of marker differences across batches. To illustrate this,
we used the same spatial dataset described in the previous section to
compare manually annotated brain layers to the donor IDs. Figure 2
explores the differences between WM in donors Br8100 and
Br5595. In this case, we see strong differences in the
normalized expression of several interesting genes. For example, we see
large differences again for MBP, a marker gene that is central to the
manual annotations. Likewise, we see batch effects in other notable
genes such as CNP, identified by Zeng et. al., 2021
as brain cortex cell-type marker genes conserved between Human and
Mouse. Finally, we see also differences for MOBP, highlighting this gene
as not only variable across layers but also across batches.
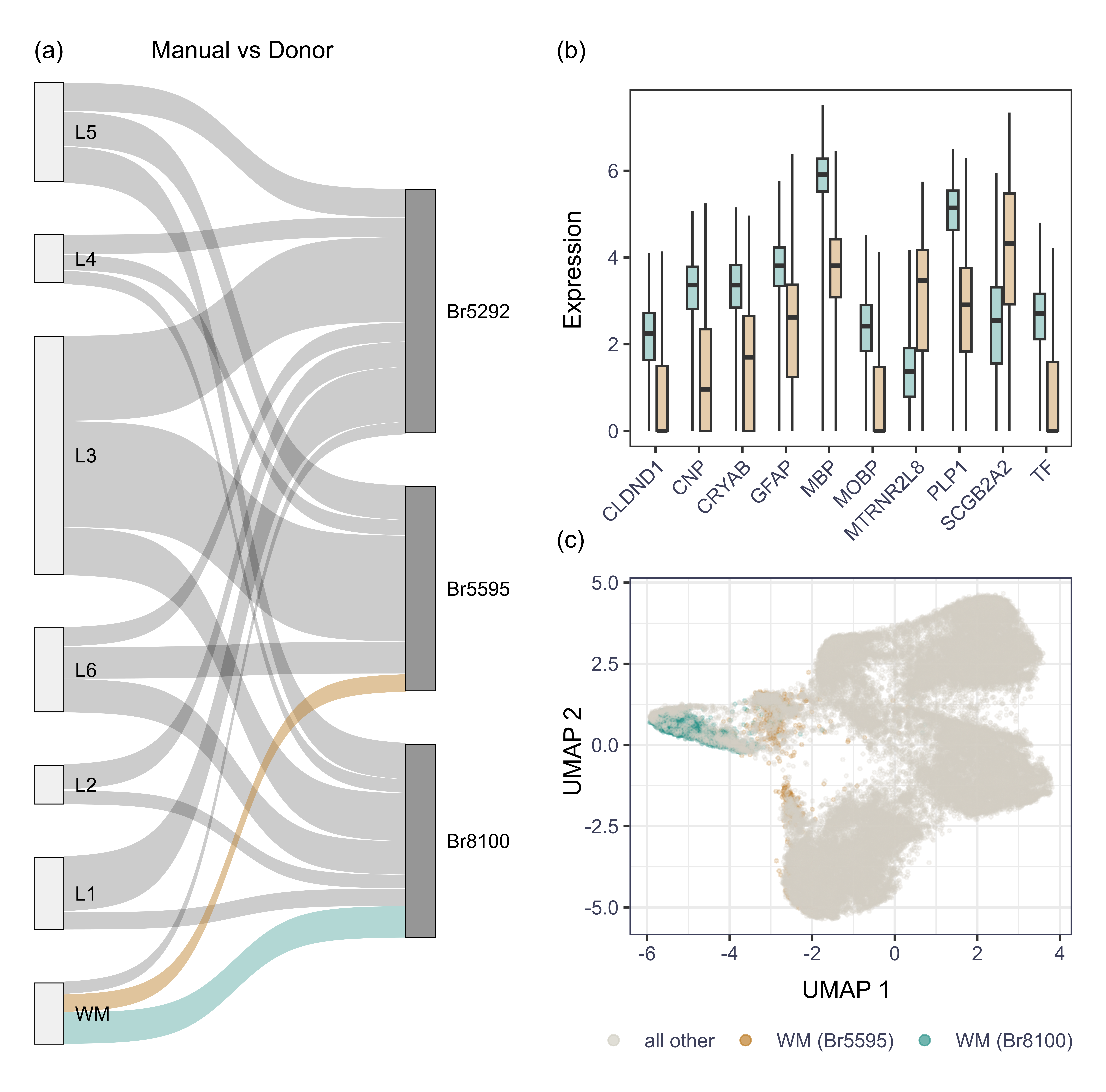
tagtango to only include in-tissue spots. The
coloured links in the diagram indicate the cell populations selected for
deeper analysis. Panel (b) displays a direct comparison of the
normalized RNA expression for the two selected cell populations,
including only markers selected as relevant. The colours of the bars
match those of the selected links in panel (a). Panel (c) presents the
UMAP representation of the RNA expression for all spots, where the
colours of the points match those of the selected links in panel
(a).Supplementary usage scenario: comparing single-cell datasets
As the final usage case scenario, we showcased how
tagtango can be used to compare datasets. To do so, we used
two independent 10x datasets: PBMCs from a healthy donor obtained by 10x
Genomics from AllCells (10x
Genomics, 2021), and PBMCs form a diseased Acute Lymphoblastic
Leukemia donor obtained by 10x Genomics from Sanguine Biosciences (10x
Genomics, 2024).
In order to analyse the datasets together with tagtango,
we first independently processed and annotated each of them. To do so,
we followed a three-step process: first, we downloaded the corresponding
h5 file from the 10x Genomics platform; we then processed each dataset
using the R packages Seurat (Hao et. al.,
2023), SingleCellExperiment (Amezquita et. al.,
2020) and scater (McCarthy et. al.,
2017); and we use the R packages celldex and
SingleR (Liu et. al., 2019)
to annotate the single-cell datasets, identifying the main cell types.
In R, the processing of each dataset is as follows:}
# Load libraries
library(Seurat)
library(SingleCellExperiment)
library(scater)
library(SingleR)
library(celldex)
# Set path to the h5 file downloaded from www.10xgenomics.com
path <- "path_to_h5_file"
# Load raw data
sce <- as.SingleCellExperiment(
CreateSeuratObject(
Seurat::Read10X_h5(path)
)
)
# calculate the proportion of mitochondrial reads
mt.genes <- rownames(sce)[grep("^MT-",rownames(sce))]
sce <- addPerCellQC(sce, subsets = list(Mito = mt.genes))
# perform QC
qc.lib <- isOutlier(sce$sum, log=TRUE, type="lower")
qc.nexprs <- isOutlier(sce$detected, log=TRUE, type="lower")
qc.mito <- isOutlier(sce$subsets_Mito_percent, type="higher")
sce$discard <- qc.lib | qc.nexprs | qc.mito
sce <- sce[,!sce$discard]
# Normalize counts
sce <- computeLibraryFactors(sce)
sce <- logNormCounts(sce)
# Annotate dataset with SingleR
ref <- BlueprintEncodeData()
pred <- SingleR(test=sce, ref=ref, labels=ref$label.main)
colData(sce) <- cbind(colData(sce), Main.labels = pred$labels)This workflow provided us with two annotated
SingleCellExperiment, one for the healthy patient
(i.e.~sce\_healthy) and one for the cancer patient
(i.e.~sce\_cancer). The last step before analysing the
files with tagtango was to integrate these objects,
removing biologically irrelevant batch effects. To do so, we used
packages scran (Lun et. al.,
2016) and batchelor (Haghverdi et. al., 2018) to
correct the log-expression values via linear regression:
# Load libraries
library(scran)
library(batchelor)
# Find genes in common
universe <- intersect(rownames(sce_cancer_clean), rownames(sce_healthy_clean))
# Variance model
dec_cancer_clean <- modelGeneVarByPoisson(sce_cancer)[universe,]
dec_healthy_clean <- modelGeneVarByPoisson(sce_healthy)[universe,]
# Find HVGs
combined.dec <- combineVar(dec_cancer_clean, dec_healthy_clean)
chosen.hvgs <- combined.dec$bio > 0
# Per-batch scaling normalization
rescaled <- multiBatchNorm(sce_cancer[universe,], sce_healthy[universe,])
pbmc_cancer <- rescaled[[1]]
pbmc_healthy <- rescaled[[2]]
# Merge datasets and calculate TSNA
rescaled <- rescaleBatches(pbmc_cancer, pbmc_healthy)
rescaled <- runPCA(rescaled, subset_row=chosen.hvgs,
exprs_values="corrected",
BSPARAM=BiocSingular::RandomParam())
rescaled <- runTSNE(rescaled, dimred="PCA")
# Copy annotations and batch information
rescaled$batch <- factor(rescaled$batch, levels = c(1,2), labels = c("cancer", "healthy"))
rescaled$annotations <- c(pbmc_cancer$Main.labels, pbmc_healthy$Main.labels)Using tagtango, we then compared the batch information
and the annotations found with singleR (Figure 3). In
particular, we focused on the differences across cells annotated as CD4+
T-cells across the two datasets. Again, tagtango
highlighted differences in the normalized expression of several
interesting genes, all related to immune response in cancer. Notably,
JUNB has been identified in the past as a gatekeeper for a certain type
of lymphoid leukemia (Ott et. al., 2007),
and LTB has been shown to promote the development of T-cell acute
lymphoblastic leukemia (Fernandes et. al., 2015).
Likewise, genes such as CD27 or IL-32 have been closely associated to
the clinical outcome and prognostic of acute lymphoblastic leukemia
patients (Chen et. al.,
2017; Abobakr
et. al., 2023; Shim et. al.,
2022), and there are links between the expression of IL-7R and TCF7
with subsets of these patients (Oliveira et. al.,
2019; Van
Thillo et. al., 2021). Overall, tagtango was able to
quickly identify genes that are associated with lymphocyte development,
transcriptional regulation, and immune signaling pathways in acute
lymphoblastic leukemia patients. However, further work would be required
to validate these findings and ensure their biological relevance.
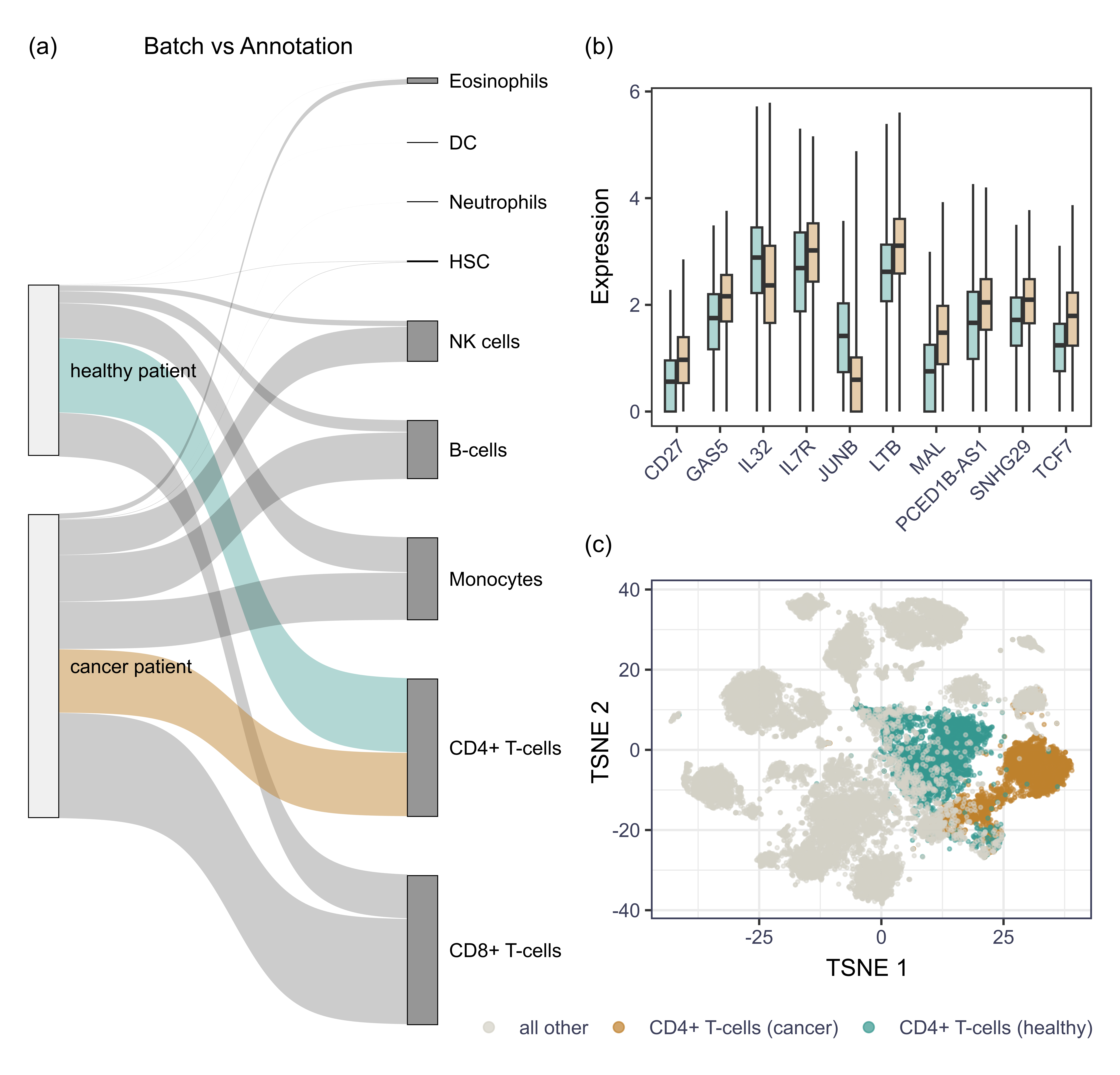
tagtango. Panel (a) displays a Sankey
diagram comparing the batch information and main cell types. The
coloured links in the diagram indicate the cell populations selected for
deeper analysis. Panel (b) displays a direct comparison of the
normalized and batch-corrected RNA expression for the two selected cell
populations, including only markers selected as relevant. The colours of
the bars match those of the selected links in panel (a). Panel (c)
presents the TSNE representation of the RNA expression, where the
colours of the points match those of the selected links in panel
(a).